|
AnnoDUF
AnnoDUF is a comprehensive database for DUFs that also offers algorithms to provide
putative annotations for newly identified protein sequences. This can enhance understanding of biological processes and aid in
drug discovery. The homepage features a brief overview of the database content and its algorithms.
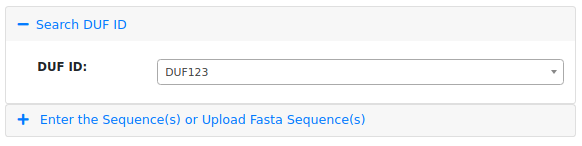
The figure above shows that users can select a DUF ID by clicking on the "Select" option and choosing the DUF ID with their research interest.
For example, a DUF ID "DUF123" is been selected for the analysis
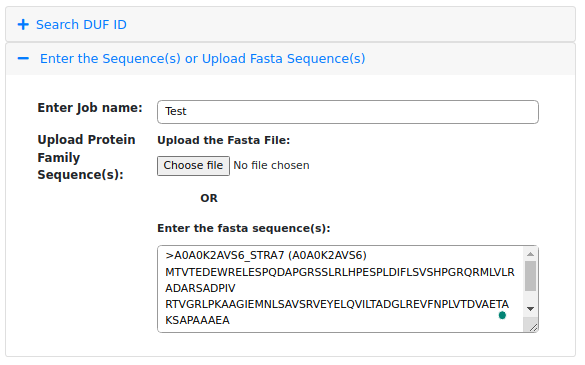
The figure above shows that users can upload a FASTA file by clicking on the "Choose File" option, or
they can directly submit the FASTA sequence in the text area provided. For example, a FASTA sequence of
a PD-(D/E)XK motif protein has been submitted in the text area.

The figure above shows that users can run an example by clicking on the "Example" button. After selecting
the DUF ID, uploading a FASTA file, or submitting the FASTA sequence of the protein of interest
in the text area, users should click on the "Analyze" button to identify the putative annotation of the
protein. Additionally, users can reset all fields by clicking on the "Reset" button.
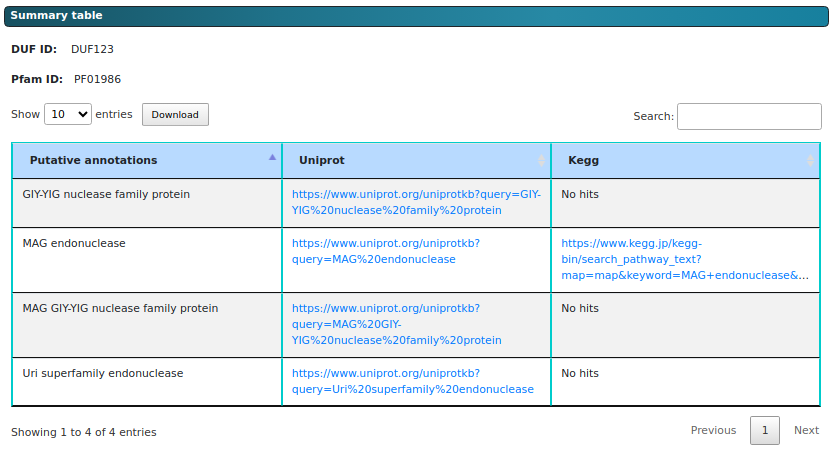
The figure above shows the summary table that users will receive after submitting their query and
completing the analysis. This interactive table allows users to search for keywords using the search box
located at the top right corner of the table. Users can also download the table by clicking the
"Download" button at the top left corner. Additionally, pagination options are available at the bottom
right corner, enabling easier navigation throughout the table.
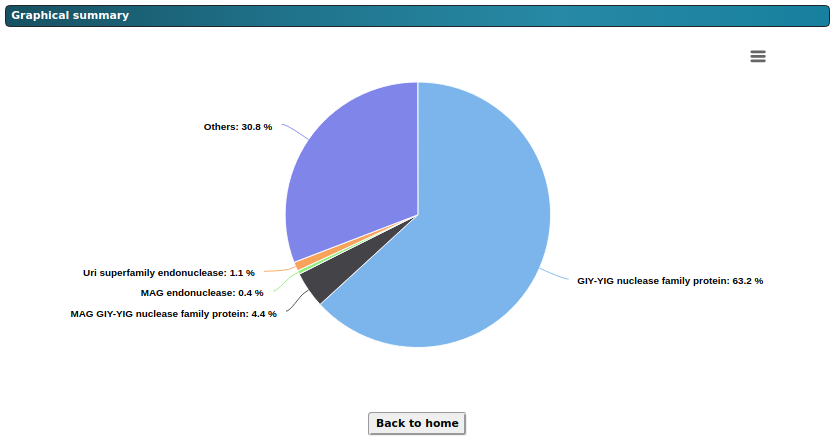
The figure above shows an interactive graphical summary that visually represents the distribution of
domains within the DUFs using a pie chart. Each segment of the pie chart indicates the percentage of
distinct domains found in the DUF sequence collection. The images are available for download in .png,
.jpeg, .pdf, and .svg formats, and the data used to create the pie chart is also downloadable.
Additionally, clicking on "Reset to Home" will redirect users to the AnnoDUF homepage.
|
|
X-Pro
X-Pro is an advanced computational tool designed to analyze potential rotameric
configurations resulting from point mutations in protein structures by leveraging two readily accessible
used software tools, PyMOL and LigPlot, X-Pro streamlines the process of studying mutations and their
structural implications.
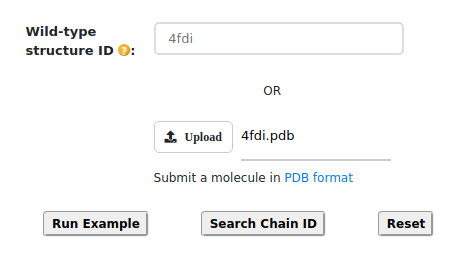
The figure above illustrates two methods for providing the wild-type structure: users can either upload
the wild-type structure directly or enter a PDB ID, allowing the structure to be retrieved from the
Protein Data Bank Database. For instance, the PDB ID "4FDI" is been selected for analysis. Additionally,
users can reset the wild-type structure by clicking the "Reset" button or run a predefined example using
the "Run Example" button.
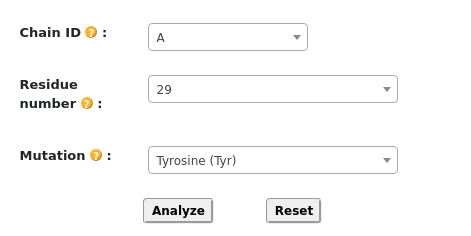
Once the wild-type structure is submitted, the mutation detail options become dynamically accessible. As
shown in the figure, users can specify mutation details, including the residue ID, chain ID, and the
desired amino acid mutation. Furthermore, all fields can be cleared by clicking the "Reset" button.

The above figure summarizes details of the mutational analysis entered by the user which includes the
wild-type structure ID, chain ID, residue ID, the desired amino acid mutation, and also the potential
rotamers identified during the mutation process using PyMOL.
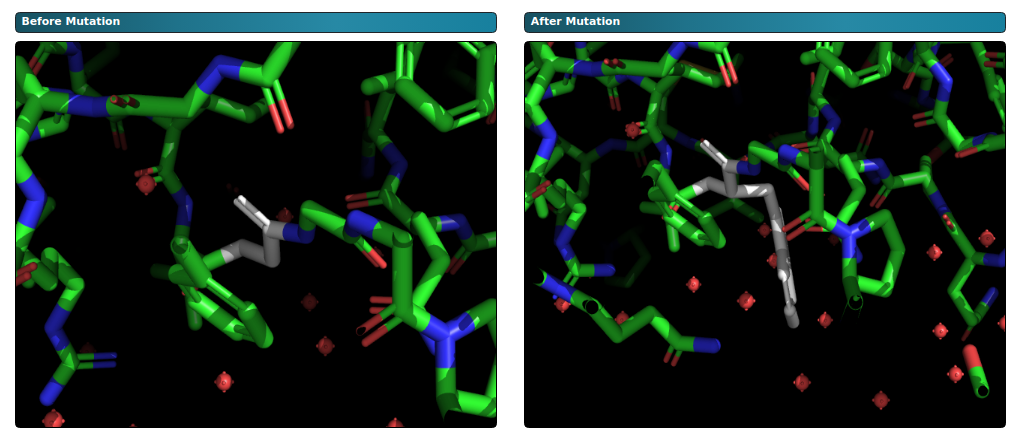
The above figure shows the graphical representation generated by PyMOL which illustrates the mutation within
the enzyme. For instance, glycine at position 115 in chain A is substituted with tryptophan.
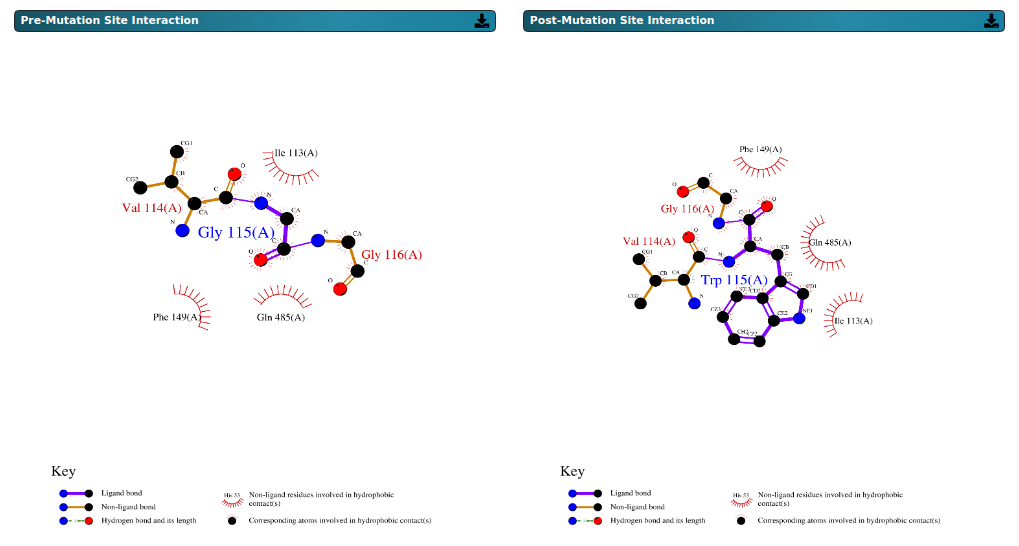
The above figure represents the LigPlot+ visualization which highlights the interactions at the mutation
site before and after the substitution. For example, in the pre-mutation state, the ligand forms a bond
with glycine at position 116. Following the mutation, the post-mutation interaction shifts to a
hydrophobic contact involving glycine at position 116, as depicted in the post-mutation state figure.
The images are available for download in .png format.
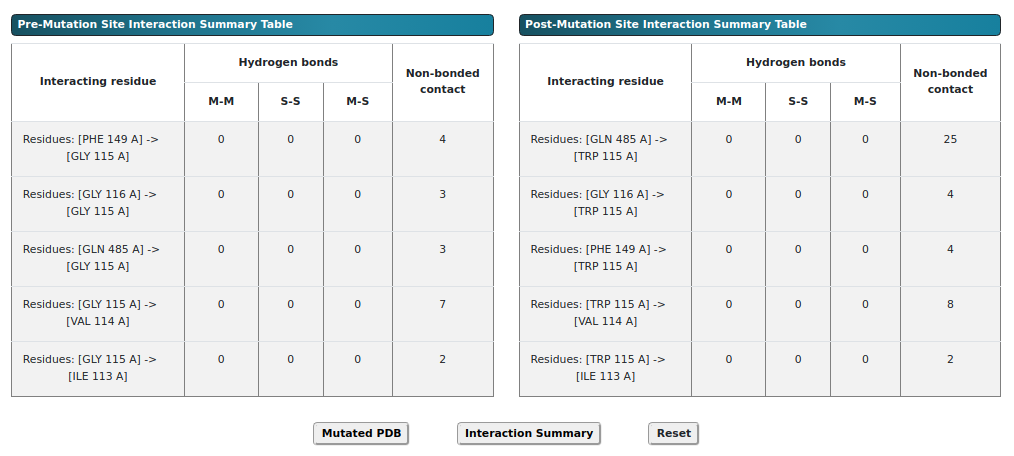
The figure above presents a summary table comparing the pre-mutation and post-mutation interactions of
the enzyme. The table includes details such as the number of residues interacting with glycine, the
number of hydrogen bonds formed, and the non-bonded contacts, including hydrophobic interactions. In the
table, "M" denotes interactions involving the main chain, while "S" indicates those involving the side
chain of the protein. It also highlights key interaction types, such as hydrogen bonds (non-covalent)
and electrostatic interactions.
Users can download the interaction summary table by clicking the "Interaction Summary" button located at
the end. Additionally, the mutated protein structure is available for download, allowing for further
mutational analysis.
|